Nous avenços en la recerca de la síndrome de Schaaf-Yang
Un equip liderat per investigadores de l'Institut de Recerca Sant Joan de Déu · IBUB descobreixen que les mutacions del gen MAGEL2 generen proteïnes truncades no funcionals que tendeixen a acumular-se en el nucli cel·lular. Les mutacions del gen MAGEL2 causen la síndrome de Schaaf-Yang (SYS), una malaltia ultrarara que afecta el desenvolupament neuronal i cognitiu dels infants.
Els nous avenços en la recerca de la SYS els publica ara un estudi de la revista Journal of Medical Genetics liderat per un equip de la Facultat de Biologia i de lʼInstitut de Biomedicina de la Universitat de Barcelona (IBUB), lʼInstitut de Recerca Sant Joan de Déu (IRSJD) i el Centre dʼInvestigació Biomèdica en Xarxa de Malalties Rares (CIBERER). Cal recordar que aquest equip també és autor de la publicació de la primera guia clínica sobre la síndrome de Schaaf-Yang (Journal of Medical Genetics, 2022), adreçada a professionals sanitaris i famílies dʼinfants afectats per aquesta patologia.
Comprendre millor la funció, les variants genètiques i l'impacte de la retenció nuclear de la proteïna MAGEL2 obrirà noves vies per dissenyar teràpies genètiques específiques dirigides a pacients a fi d'evitar la síntesi de la proteïna alterada i abordar la SYS, una malaltia encara sense cura o tractament.
Mutacions genètiques que originen proteïnes truncades
El gen MAGEL2 es troba al cromosoma 15, s'expressa especialment en el sistema nerviós i produeix la proteïna MAGEL2, implicada en el transport retrògrad i el reciclatge de proteïnes en el citoplasma cel·lular de les neurones. Fins ara, s'han documentat en la bibliografia científica més de vuitanta mutacions en el gen MAGEL2, algunes de les quals es troben repetides entre pacients. Actualment, es calcula que hi ha prop de 250 persones diagnosticades amb la síndrome de Schaaf-Yang arreu del món.
El nou treball, fet amb cèl·lules humanes in vitro, constata com gairebé totes les proteïnes truncades associades amb la síndrome de Schaaf-Yang perden una part de l'estructura molecular a causa de les mutacions genètiques.
“Les proteïnes MAGEL2 funcionals tenen una estructura molecular completa que els permet interactuar amb altres proteïnes i dur a terme les seves funcions biològiques normals. Solen trobar-se en ubicacions específiques dins el citoplasma de la cèl·lula, principalment en compartiments subcel·lulars relacionats amb el transport i el reciclatge de proteïnes”, detalla la Dra. Susanna Balcells, coordinadora del grup de recerca Genètica Molecular Humana I de l’IRSJD. “En canvi -continua- les proteïnes truncades són versions més curtes de la proteïna MAGEL2, ja que s'han vist afectades per mutacions genètiques. Per tant, a les proteïnes truncades els manquen certes regions necessàries per funcionar correctament en la cèl·lula”.
A causa de les mutacions genètiques, les proteïnes trucades perden dominis estructurals clau, com ara el domini d'homologia MAGE, que és crucial per a les interaccions amb altres proteïnes.
“L'absència d'aquest domini podria impedir aquestes interaccions essencials per al funcionament correcte de MAGEL2, com el seu paper en el transport retrògrad i el reciclatge de proteïnes”, explica la Dra. Roser Urreizti, investigadora del grup Metabolòmica i genètica bioquímica.
Quan les proteïnes tòxiques s'acumulen al nucli de la cèl·lula
Les proteïnes truncades solen acumular-se dins del nucli cel·lular i això podria agreujar encara més la simptomatologia dels afectats per la Schaaf-Yang.
“És probable que en un context cel·lular real una part de les proteïnes truncades sintetitzades es puguin trobar també en ubicacions específiques dins del citoplasma, com, per exemple, els endosomes. Ara bé, com que la seva estructura està alterada, potser no serien capaces de fer les seves funcions normals correctament”. Tal com detalla Mónica Centeno, primera autora de l’article i investigadora del grup Genètica Molecular Humana I.
La gravetat de les manifestacions clíniques en els afectats per la síndrome de Schaaf-Yang es podria relacionar amb l'acumulació de proteïnes alterades en el nucli cel·lular. “És a dir, tot indica que les mutacions que provoquen símptomes més greus també fan que la proteïna MAGEL2 truncada s'acumuli més al nucli. Això podria explicar-se per què una acumulació superior de proteïnes truncades al nucli podria estar interferint amb processos nuclears importants i afectar en major grau el funcionament de la cèl·lula en condicions normals”, afirma la Dra. Raquel Rabionet, investigadora del grup Genètica Molecular Humana I.
A la recerca de tractaments per abordar la síndrome de Schaaf-Yang
Els mecanismes cel·lulars que impulsen la degradació de proteïnes -per exemple, el sistema ubiquitina-proteasoma- podrien ajudar a reduir els efectes negatius de les proteïnes truncades i contribuir a mitigar l'evolució de la patologia. Tanmateix, si la taxa de producció de proteïnes supera la capacitat de degradació de la cèl·lula, les proteïnes aberrants podrien escapar a la degradació i continuar exercint efectes tòxics.
“En aquest sentit, hem comprovat que les proteïnes MAGEL2 normals i truncades tenen una vida mitjana molt similar. Per tant, la MAGEL2 truncada es trobaria estable a la cèl·lula, on podria estar exercint els efectes tòxics”, comenta Aina Prat. “Si coneguéssim com les proteïnes MAGEL2 truncades alteren la funció cel·lular, es podrien desenvolupar estratègies per promoure la degradació d'aquestes proteïnes tòxiques, restaurar la funció cel·lular o compensar les disfuncions metabòliques i de senyalització causades per la seva acumulació”, clou l'equip, que impulsarà noves recerques per contribuir al desenvolupament de tractaments innovadors per a les persones afectades per la SYS.
Article de referència
Centeno, Mónica; Alcaide-Consuegra, Estefanía; Gibson, Sophie; Prat-Planas, Aina; Gutiérrez-Ávila, Juan D.; Grinberg, Daniel; Urreizti, Roser; Rabionet, Raquel; Balcells, Susanna. «Subcellular localisation of truncated MAGEL2 proteins: insight into the molecular pathology of Schaaf-Yang syndrome». Journal of Medical Genetics, març de 2024. Doi: 10.1136/jmg-2024-109898
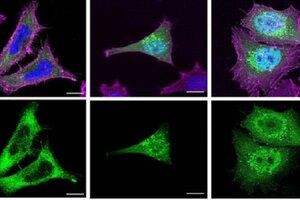
Aquesta acumulació progressiva de proteïnes anòmales, a més, podria causar un efecte tòxic en els pacients afectats per la síndrome, que estan afectats per malformacions congènites, retard intel·lectual, alteracions en els trets facials, apnea del son i contractures articulars.
